| Citation: | Guanghui Cai, Zhendong Cao, Fankai Xie, Huaxian Jia, Wei Liu, Yaxian Wang, Feng Liu, Xinguo Ren, Sheng Meng, Miao Liu. Predicting structure-dependent Hubbard U parameters via machine learning[J]. Materials Futures, 2024, 3(2): 025601. doi: 10.1088/2752-5724/ad19e2

|
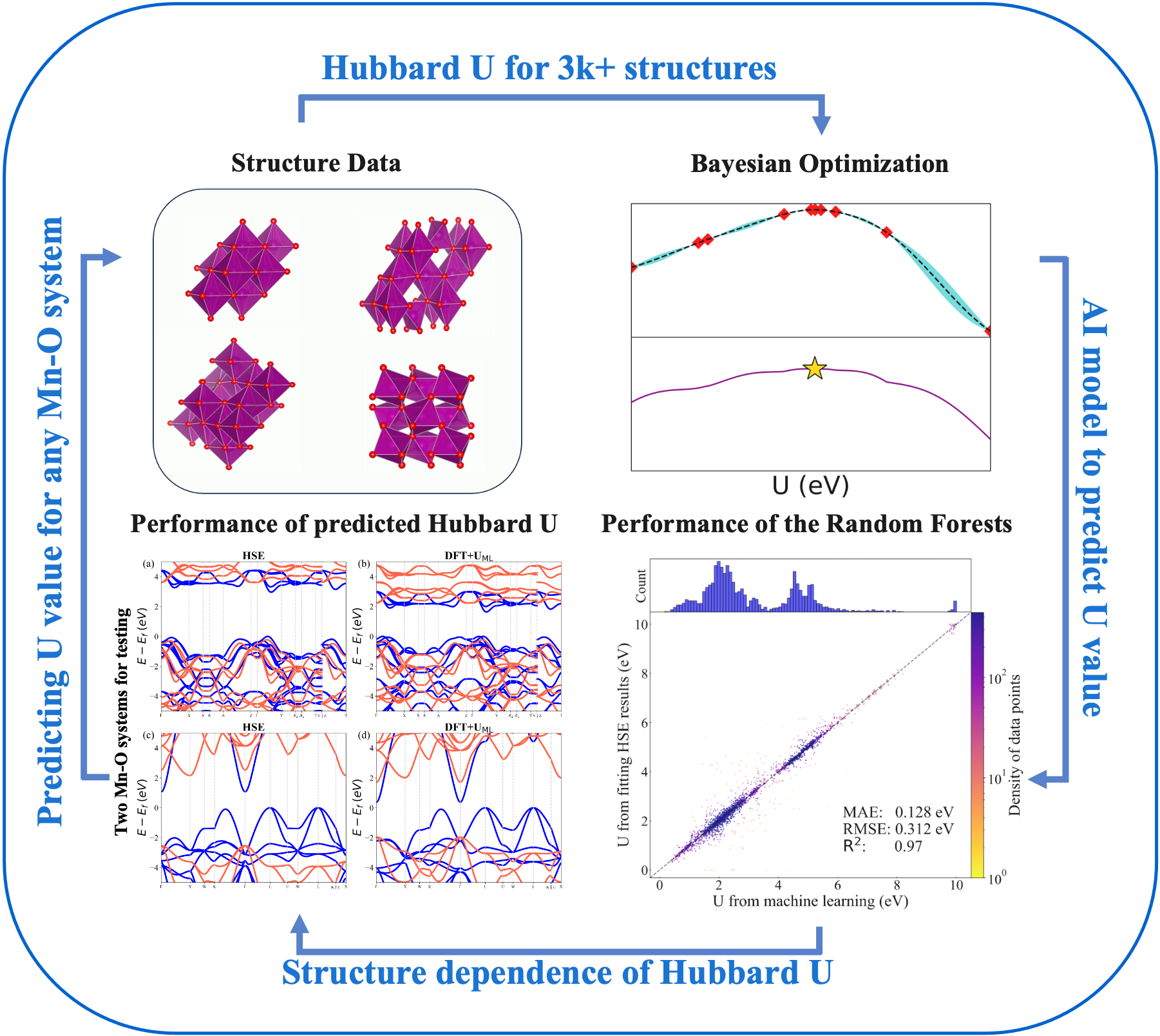
DFT + U is a widely used treatment in the density functional theory (DFT) to deal with correlated materials that contain open-shell elements, whereby the quantitative and sometimes even qualitative failures of local and semi-local approximations can be corrected without much computational overhead. However, finding appropriate U parameters for a given system and structure is non-trivial and computationally intensive, because the U value has generally a strong chemical and structural dependence. In this work, we address this issue by building a machine learning (ML) model that enables the prediction of material- and structure-specific U values at nearly no computational cost. Using Mn–O system as an example, the ML model is trained by calibrating DFT + U electronic structures with the hybrid functional results of more than 3000 structures. The model allows us to determine an accurate U value (MAE = 0.128 eV, R2 = 0.97) for any given Mn–O structure. Further analysis reveals that M–O bond lengths are key local structural properties in determining the U value. This approach of the ML U model is universally applicable, to significantly expand and solidify the use of the DFT + U method.
| [1] |
Perdew J P, Burke K and Ernzerhof M 1996 Generalized gradient approximation made simple Phys. Rev. Lett. 77 3865–8
|
| [2] |
Becke A D 1988 Density-functional exchange-energy approximation with correct asymptotic behavior Phys. Rev. A 38 3098–100
|
| [3] |
Lee C, Yang W and Parr R G 1988 Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density Phys. Rev. B 37 785–9
|
| [4] |
Anisimov V I, Zaanen J and Andersen O K 1991 Band theory and Mott insulators: Hubbard U instead of Stoner I Phys. Rev. B 44 943–54
|
| [5] |
Dudarev S L, Botton G A, Savrasov S Y, Humphreys C J and Sutton A P 1998 Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study Phys. Rev. B 57 1505–9
|
| [6] |
Han M J, Ozaki T and Yu J O 2006 (N) LDA + U electronic structure calculation method based on the nonorthogonal pseudoatomic orbital basis Phys. Rev. B 73 045110
|
| [7] |
Wang L, Maxisch T and Ceder G 2006 Oxidation energies of transition metal oxides within the GGA + U framework Phys. Rev. B 73 195107
|
| [8] |
Zhou F, Cococcioni M, Marianetti C A, Morgan D and Ceder G 2004 First-principles prediction of redox potentials in transition-metal compounds with LDA + U Phys. Rev. B 70 235121
|
| [9] |
Cococcioni M and de Gironcoli S 2005 Linear response approach to the calculation of the effective interaction parameters in the LDA + U method Phys. Rev. B 71 035105
|
| [10] |
Aryasetiawan F, Karlsson K, Jepsen O and Schönberger U 2006 Calculations of Hubbard U from first-principles Phys. Rev. B 74 125106
|
| [11] |
Miyake T and Aryasetiawan F 2008 Screened Coulomb interaction in the maximally localized Wannier basis Phys. Rev. B 77 085122
|
| [12] |
Şaşıoǧlu E, Friedrich C and Blügel S 2011 Effective Coulomb interaction in transition metals from constrained random-phase approximation Phys. Rev. B 83 121101
|
| [13] |
Yu M, Yang S, Wu C and Marom N 2020 Machine learning the Hubbard U parameter in DFT+U using Bayesian optimization npj Comput. Mater. 6 180
|
| [14] |
Yu M, Moayedpour S, Yang S, Dardzinski D, Wu C, Pribiag V S and Marom N 2021 Dependence of the electronic structure of the EuS/InAs interface on the bonding configuration Phys. Rev. Mater. 5 064606
|
| [15] |
Yang S, Dardzinski D, Hwang A, Pikulin D I, Winkler G W and Marom N 2021 First-principles feasibility assessment of a topological insulator at the InAs/GaSb interface Phys. Rev. Mater. 5 084204
|
| [16] |
Popov M N, Spitaler J, Romaner L, Bedoya-Martínez N and Hammer R 2021 Bayesian optimization of Hubbard U’s for investigating InGaN superlattices Electron. Mater. 2 370–81
|
| [17] |
Lu D and Liu P 2014 Rationalization of the Hubbard U parameter in CeOx from first principles: unveiling the role of local structure in screening J. Chem. Phys. 140 084101
|
| [18] |
Hsu H, Umemoto K, Cococcioni M and Wentzcovitch R 2009 First-principles study for low-spin LaCoO3 with a structurally consistent Hubbard U Phys. Rev. B 79 125124
|
| [19] |
Tsuchiya T, Wentzcovitch R M, da Silva C R S and de Gironcoli S 2006 Spin transition in magnesiowüstite in Earth’s lower mantle Phys. Rev. Lett. 96 198501
|
| [20] |
Kulik H J and Marzari N 2011 Accurate potential energy surfaces with a DFT+U(R) approach J. Chem. Phys. 135 194105
|
| [21] |
Heyd J, Scuseria G E and Ernzerhof M 2003 Hybrid functionals based on a screened Coulomb potential J. Chem. Phys. 118 8207–15
|
| [22] |
Heyd J, Scuseria G E and Ernzerhof M 2006 Erratum: ‘Hybrid functionals based on a screened Coulomb potential’ [J. Chem. Phys. 118, 8207 (2003)] J. Chem. Phys. 124 219906
|
| [23] |
Amit Y and Geman D 1997 Shape quantization and recognition with randomized trees Neural Comput. 9 1545–88
|
| [24] |
Ho T K 1998 The random subspace method for constructing decision forests IEEE Trans. Pattern Anal. Mach. Intell. 20 832–44
|
| [25] |
Kresse G and Furthmüller J 1996 Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set Phys. Rev. B 54 11169–86
|
| [26] |
Kresse G and Joubert D 1999 From ultrasoft pseudopotentials to the projector augmented-wave method Phys. Rev. B 59 1758–75
|
| [27] |
Blöchl P E 1994 Projector augmented-wave method Phys. Rev. B 50 17953–79
|
| [28] |
Tavadze P, Boucher R, Avendáno-Franco G, Kocan K X, Singh S, Dovale-Farelo V, Ibarra-Hernández W, Johnson M B, Mebane D S and Romero A H 2021 Exploring DFT+U parameter space with a Bayesian calibration assisted by Markov chain Monte Carlo sampling npj Comput. Mater. 7 182
|
| [29] |
Liechtenstein A I, Anisimov V I and Zaanen J 1995 Density-functional theory and strong interactions: orbital ordering in Mott-Hubbard insulators Phys. Rev. B 52 R5467–70
|
| [30] |
Blum V, Gehrke R, Hanke F, Havu P, Havu V, Ren X, Reuter K and Scheffler M 2009 Ab initio molecular simulations with numeric atom-centered orbitals Comput. Phys. Commun. 180 2175–96
|
| [31] |
Ren X, Rinke P, Blum V, Wieferink J, Tkatchenko A, Sanfilippo A, Reuter K and Scheffler M 2012 Resolution-of-identity approach to Hartree–Fock, hybrid density functionals, RPA, MP2 and GW with numeric atom-centered orbital basis functions New J. Phys. 14 053020
|
| [32] |
Levchenko S V, Ren X, Wieferink J, Johanni R, Rinke P, Blum V and Scheffler M 2015 Hybrid functionals for large periodic systems in an all-electron, numeric atom-centered basis framework Comput. Phys. Commun. 192 60–69
|
| [33] |
Ong S P, Richards W D, Jain A, Hautier G, Kocher M, Cholia S, Gunter D, Chevrier V L, Persson K A and Ceder G 2013 Python materials genomics (pymatgen): a robust, open-source python library for materials analysis Comput. Mater. Sci. 68 314–9
|
| [34] |
Fernando 2022 Bayesian optimization
|
| [35] |
Okazawa K, Tsuji Y, Kurino K, Yoshida M, Amamoto Y and Yoshizawa K 2022 Exploring the optimal alloy for nitrogen activation by combining Bayesian optimization with density functional theory calculations ACS Omega 7 45403–8
|
| [36] |
scikit-learn/scikit-learn Scikit-learn: machine learning in Python (available at: https://github.com/scikit-learn/scikitlearn)
|
| [37] |
Liang Y et al 2022 A universal model for accurately predicting the formation energy of inorganic compounds Sci. China Mater. 66 343–51
|
| [38] |
Ong S P, Wang L, Kang B and Ceder G 2008 Li−Fe−P−O 2 phase diagram from first principles calculations Chem. Mater. 20 1798–807
|
| [39] |
Jain A, Hautier G, Ong S P, Moore C J, Fischer C C, Persson K A and Ceder G 2011 Formation enthalpies by mixing GGA and GGA + U calculations Phys. Rev. B 84 045115
|
| [40] |
Liu M and Meng S 2022 Atomly.net materials database and its application in inorganic chemistry Sci. Sin.-Chim. 53 19–25
|
| [41] |
Tomczak J M, Miyake T and Aryasetiawan F 2010 Realistic many-body models for manganese monoxide under pressure Phys. Rev. B 81 115116
|
| [42] |
Ivashko O et al 2019 Strain-engineering Mott-insulating La2CuO4 Nat. Commun. 10 786
|
| [43] |
Hedin L 1965 New method for calculating the one-particle Green’s function with application to the electron-gas problem Phys. Rev. 139 A796–823
|
| [44] |
Purvis G D and Bartlett R J 1982 A full coupled-cluster singles and doubles model: the inclusion of disconnected triples J. Chem. Phys. 76 1910–8
|
| [45] |
Huhn W P and Blum V 2017 One-hundred-three compound band-structure benchmark of post-self-consistent spin-orbit coupling treatments in density functional theory Phys. Rev. Mater. 1 033803
|
| [46] |
Ye L-H, Luo N, Peng L-M, Weinert M and Freeman A J 2013 Dielectric constant of NiO and LDA + U Phys. Rev. B 87 075115
|
| [47] |
Pask J E, Singh D J, Mazin I I, Hellberg C S and Kortus J 2001 Structural, electronic, and magnetic properties of MnO Phys. Rev. B 64 024403
|
| [48] |
Deng H-X, Li J, Li S-S, Xia J-B, Walsh A and Wei S-H 2010 Origin of antiferromagnetism in CoO: a density functional theory study Appl. Phys. Lett. 96 162508
|
| [49] |
Dufek P, Blaha P, Sliwko V and Schwarz K 1994 Generalized-gradient-approximation description of band splittings in transition-metal oxides and fluorides Phys. Rev. B 49 10170–5
|
| [50] |
Jain A et al 2013 Commentary: the materials project: a materials genome approach to accelerating materials innovation APL Mater. 1 011002
|
| [51] |
Sit P H-L, Cococcioni M and Marzari N 2006 Realistic quantitative descriptions of electron transfer reactions: diabatic free-energy surfaces from first-principles molecular dynamics Phys. Rev. Lett. 97 028303
|
| [52] |
Sit P H-L, Cococcioni M and Marzari N 2007 Car–Parrinello molecular dynamics in the DFT+U formalism: structure and energetics of solvated ferrous and ferric ions J. Electroanal. Chem. 607 107–12
|
| [53] |
Leiria Campo V Jr and Cococcioni M 2010 Extended DFT + U + V method with on-site and inter-site electronic interactions J. Phys.: Condens. Matter 22 055602
|
| [54] |
Yu W et al 2023 Active learning the high-dimensional transferable Hubbard U and V parameters in the DFT + U + V scheme J. Chem. Theory. Comput. 19 6425–33
|
Figures(1)


Copyright © 2021 Materials Futures Editorial Office
Supported by:
Beijing Renhe Information Technology Co., Ltd.




 DownLoad:
DownLoad: