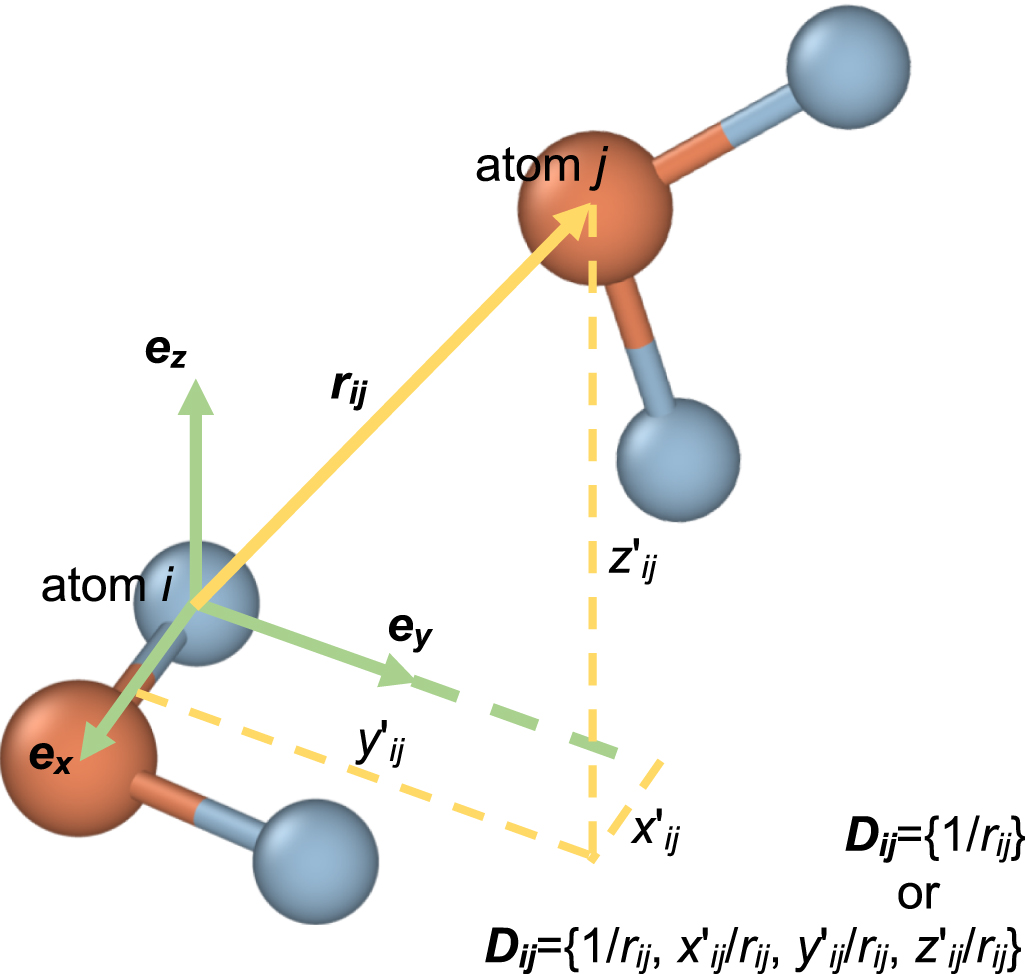
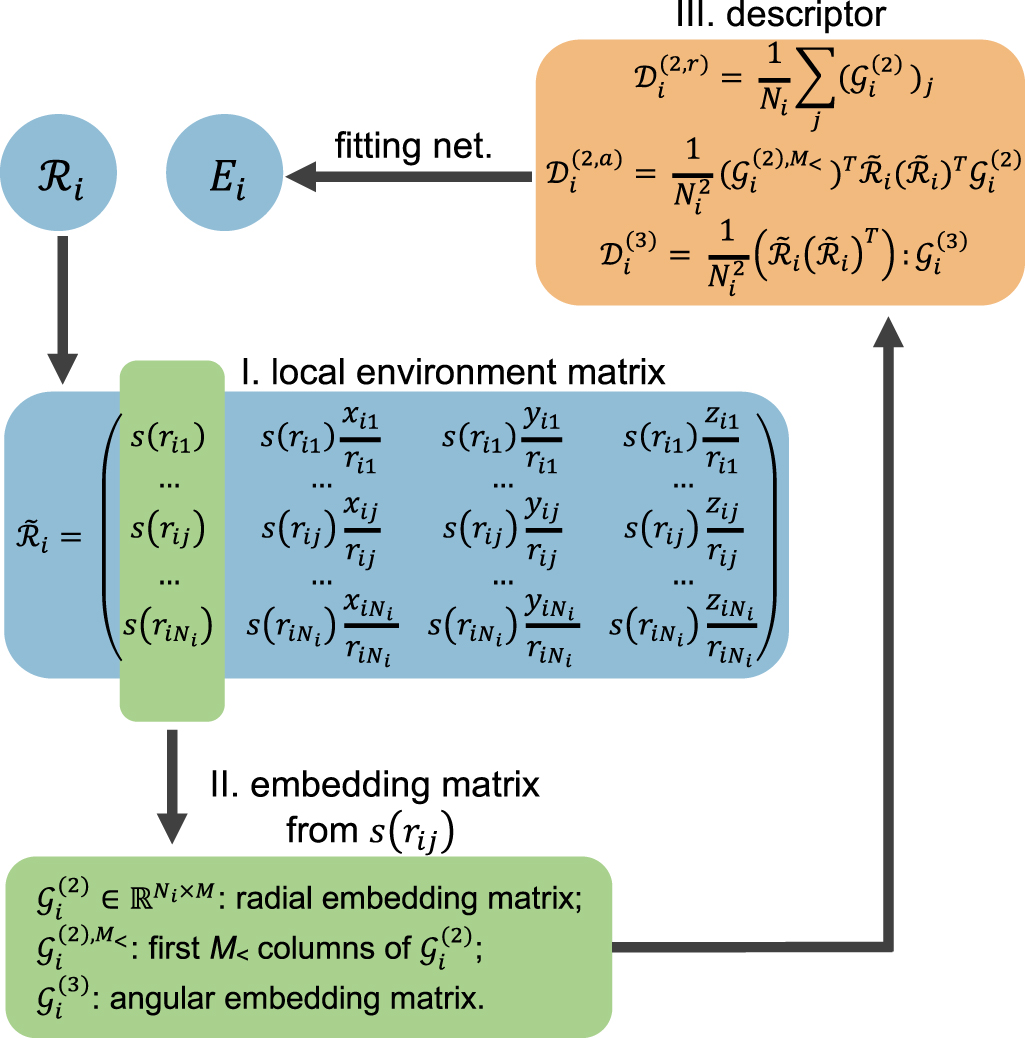
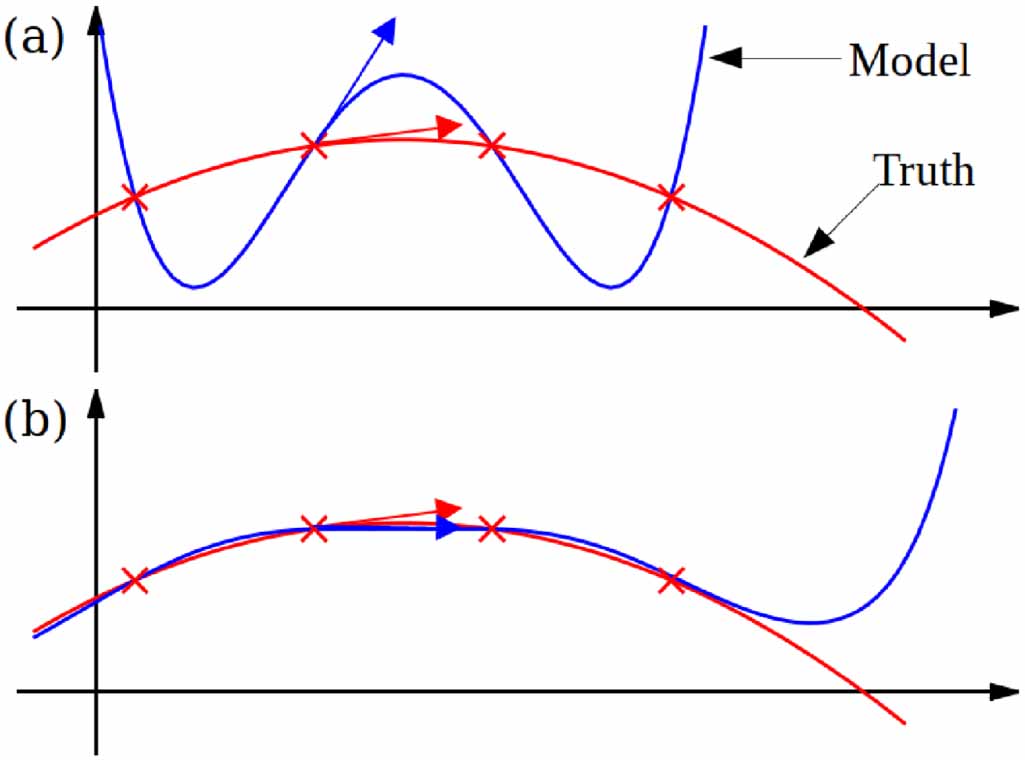
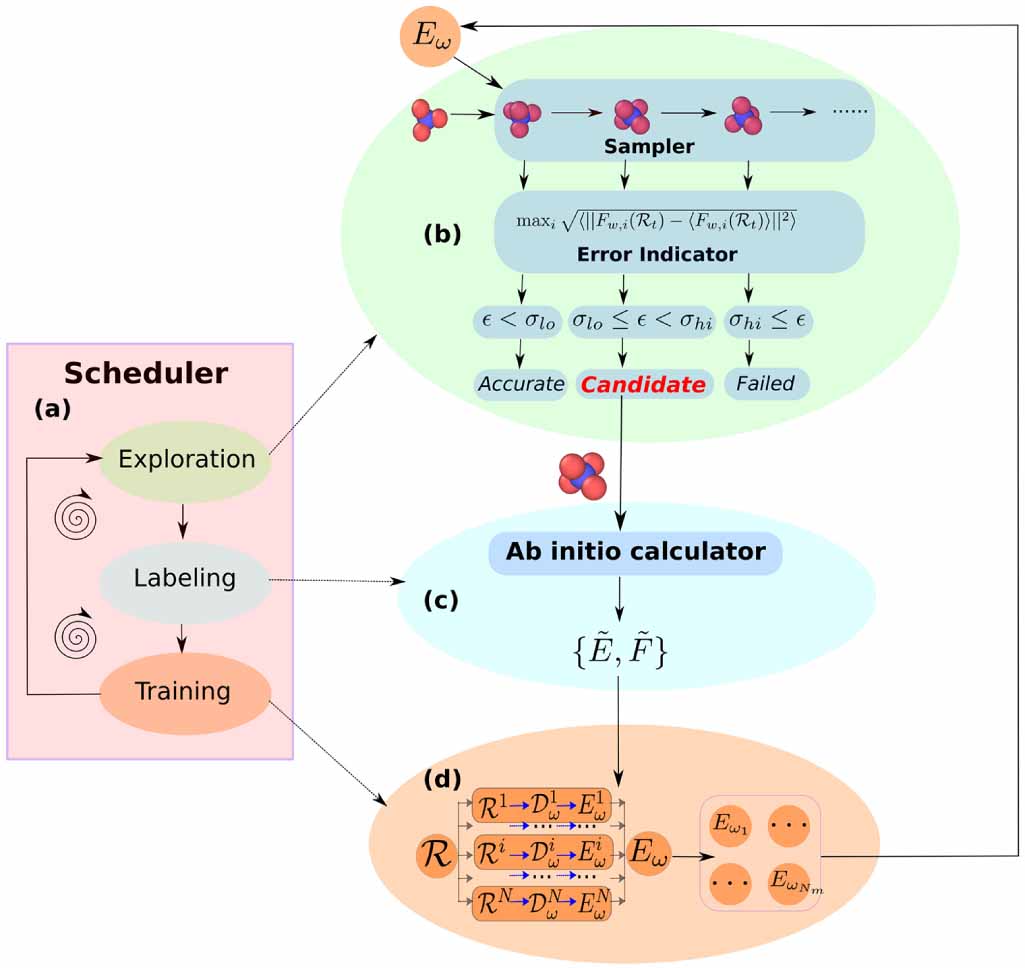
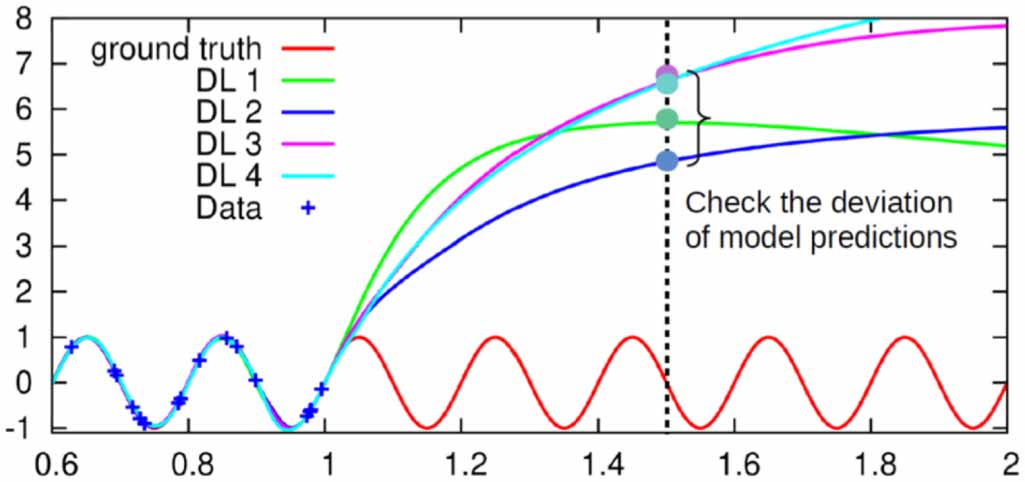
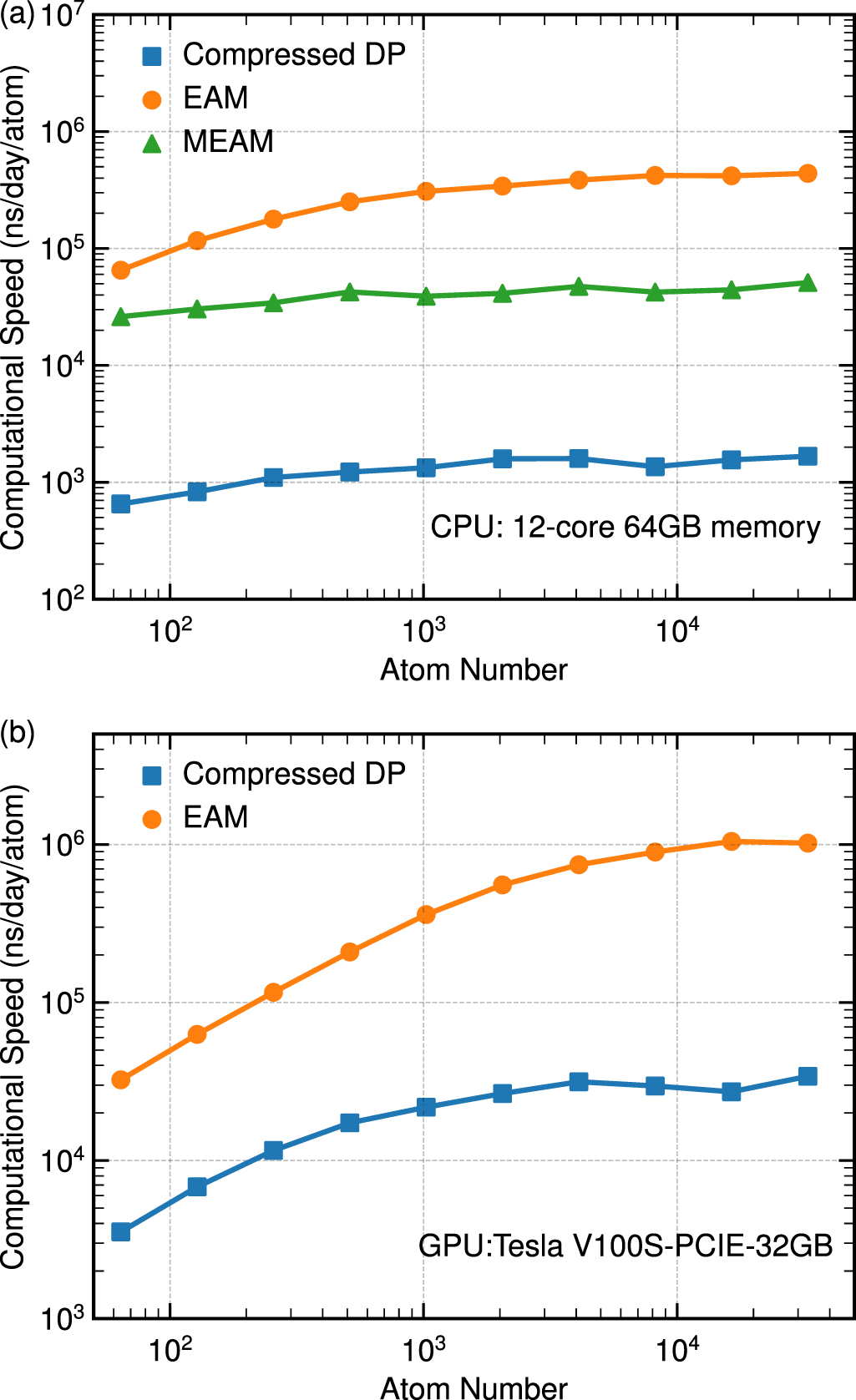
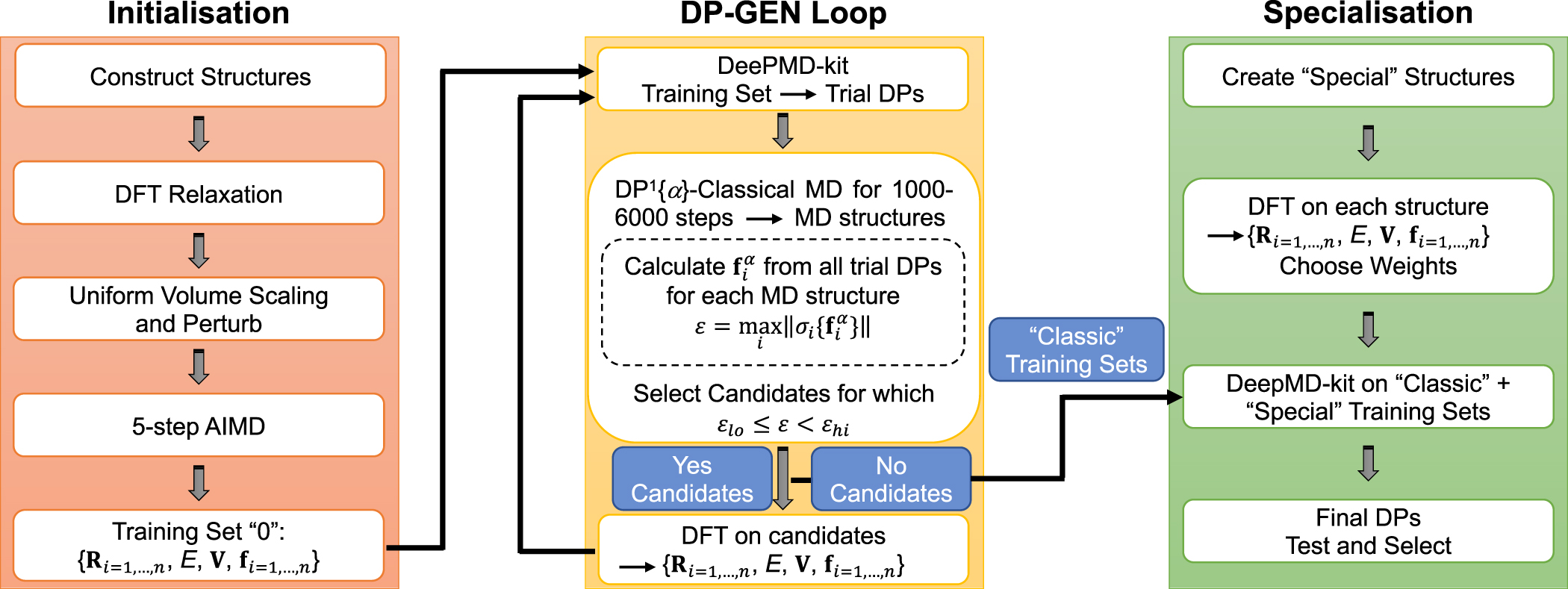
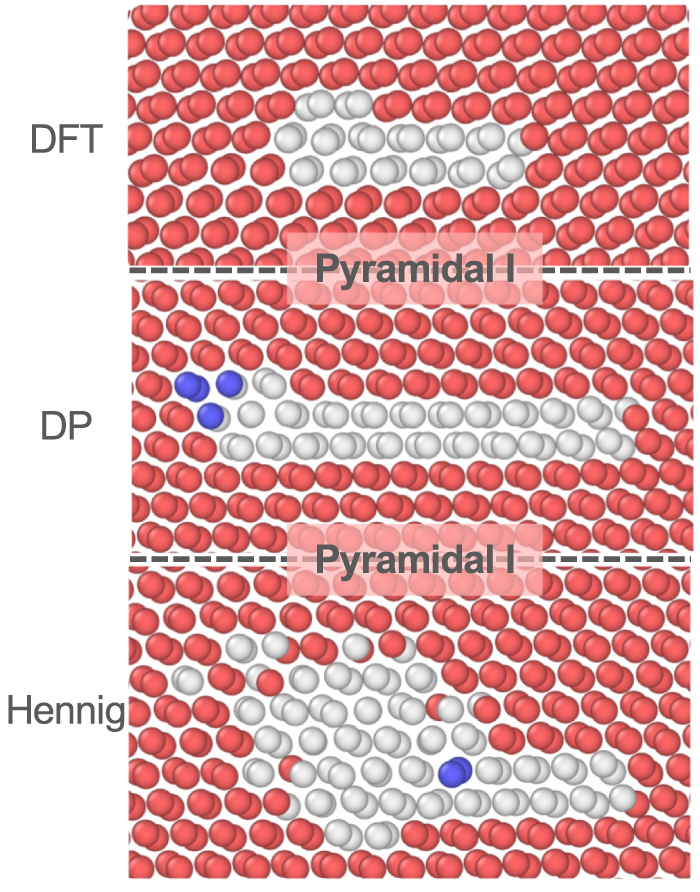
| Citation: | Tongqi Wen, Linfeng Zhang, Han Wang, Weinan E, David J Srolovitz. Deep potentials for materials science[J]. Materials Futures, 2022, 1(2): 022601. doi: 10.1088/2752-5724/ac681d

|

| [1] |
Hafner J 2000 Atomic-scale computational materials science Acta Mater. 48 71 doi: 10.1016/S1359-6454(99)00288-8
|
| [2] |
Born M, Oppenheimer R 1927 Zur quantentheorie der molekeln Ann. Phys., Lpz. 389 457 doi: 10.1002/andp.19273892002
|
| [3] |
Dirac P A M 1929 Quantum mechanics of many-electron systems Proc. R. Soc. A 123 714 doi: 10.1098/rspa.1929.0094
|
| [4] |
Kohn W, Sham L J 1965 Self-consistent equations including exchange and correlation effects Phys. Rev. 140 A1133 doi: 10.1103/PhysRev.140.A1133
|
| [5] |
Verlet L 1967 Computer experiments on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules Phys. Rev. 159 98 doi: 10.1103/PhysRev.159.98
|
| [6] |
Zwanzig R W 1954 High-temperature equation of state by a perturbation method. I. Nonpolar gases J. Chem. Phys. 22 1420 doi: 10.1063/1.1740409
|
| [7] |
Tersoff J 1989 Modeling solid-state chemistry: interatomic potentials for multicomponent systems Phys. Rev. B 39 5566 doi: 10.1103/PhysRevB.39.5566
|
| [8] |
Vink R, Barkema G, van der Weg W, Mousseau N 2001 Fitting the Stillinger-Weber potential to amorphous silicon J. Non-Cryst. Solids 282 248 doi: 10.1016/S0022-3093(01)00342-8
|
| [9] |
Daw M S, Baskes M I 1984 Embedded-atom method: derivation and application to impurities, surfaces and other defects in metals Phys. Rev. B 29 6443 doi: 10.1103/PhysRevB.29.6443
|
| [10] |
Baskes M I 1992 Modified embedded-atom potentials for cubic materials and impurities Phys. Rev. B 46 2727 doi: 10.1103/PhysRevB.46.2727
|
| [11] |
Prentice J C A, et al 2020 The ONETEP linear-scaling density functional theory program J. Chem. Phys. 152 174111 doi: 10.1063/5.0004445
|
| [12] |
Hacene M, Anciaux-Sedrakian A, Rozanska X, Klahr D, Guignon T, Fleurat-Lessard P 2012 Accelerating VASP electronic structure calculations using graphic processing units J. Comput. Chem. 33 2581 doi: 10.1002/jcc.23096
|
| [13] |
Hutchinson M, Widom M 2012 VASP on a GPU: application to exact-exchange calculations of the stability of elemental boron Comput. Phys. Commun. 183 1422 doi: 10.1016/j.cpc.2012.02.017
|
| [14] |
Jia W, Cao Z, Wang L, Fu J, Chi X, Gao W, Wang L-W 2013 The analysis of a plane wave pseudopotential density functional theory code on a GPU machine Comput. Phys. Commun. 184 9 doi: 10.1016/j.cpc.2012.08.002
|
| [15] |
Jia W, Fu J, Cao Z, Wang L, Chi X, Gao W, Wang L-W 2013 Fast plane wave density functional theory molecular dynamics calculations on multi-GPU machines J. Comput. Phys. 251 102 doi: 10.1016/j.jcp.2013.05.005
|
| [16] |
Bishop C M 2006 Pattern Recognition and Machine LearningNew YorkSpringer
|
| [17] |
Jordan M I, Mitchell T M 2015 Machine learning: trends, perspectives and prospects Science 349 255 doi: 10.1126/science.aaa8415
|
| [18] |
Mahesh B 2020 Machine learning algorithms-a review Int. J. Sci. Res. 9 381 doi: 10.21275/ART20203995
|
| [19] |
Blank T B, Brown S D, Calhoun A W, Doren D J 1995 Neural network models of potential energy surfaces J. Chem. Phys. 103 4129 doi: 10.1063/1.469597
|
| [20] |
Behler J, Parrinello M 2007 Generalized neural-network representation of high-dimensional potential-energy surfaces Phys. Rev. Lett. 98 146401 doi: 10.1103/PhysRevLett.98.146401
|
| [21] |
Khaliullin R Z, Eshet H, Khne T D, Behler J, Parrinello M 2011 Nucleation mechanism for the direct graphite-to-diamond phase transition Nat. Mater. 10 693 doi: 10.1038/nmat3078
|
| [22] |
Artrith N, Urban A 2016 An implementation of artificial neural-network potentials for atomistic materials simulations: performance for TiO2 Comput. Mater. Sci. 114 135 doi: 10.1016/j.commatsci.2015.11.047
|
| [23] |
Behler J 2021 Four generations of high-dimensional neural network potentials Chem. Rev. 121 10037 doi: 10.1021/acs.chemrev.0c00868
|
| [24] |
Behler J 2016 Perspective: machine learning potentials for atomistic simulations J. Chem. Phys. 145 170901 doi: 10.1063/1.4966192
|
| [25] |
Behler J 2017 First principles neural network potentials for reactive simulations of large molecular and condensed systems Angew. Chem., Int. Ed. 56 12828 doi: 10.1002/anie.201703114
|
| [26] |
Schtt K T, Sauceda H E, Kindermans P-J, Tkatchenko A, Mller K-R 2018 SchNeta deep learning architecture for molecules and materials J. Chem. Phys. 148 241722 doi: 10.1063/1.5019779
|
| [27] |
Schtt K T, Kessel P, Gastegger M, Nicoli K A, Tkatchenko A, Mller K-R 2019 SchNetPack: a deep learning toolbox for atomistic systems J. Chem. Theory Comput. 15 448 doi: 10.1021/acs.jctc.8b00908
|
| [28] |
Ghasemi S A, Hofstetter A, Saha S, Goedecker S 2015 Interatomic potentials for ionic systems with density functional accuracy based on charge densities obtained by a neural network Phys. Rev. B 92 045131 doi: 10.1103/PhysRevB.92.045131
|
| [29] |
Hy T S, Trivedi S, Pan H, Anderson B M, Kondor R 2018 Predicting molecular properties with covariant compositional networks J. Chem. Phys. 148 241745 doi: 10.1063/1.5024797
|
| [30] |
Unke O T, Meuwly M 2019 Physnet: a neural network for predicting energies, forces, dipole moments and partial charges J. Chem. Theory Comput. 15 3678 doi: 10.1021/acs.jctc.9b00181
|
| [31] |
Purja Pun G P, Batra R, Ramprasad R, Mishin Y 2019 Physically informed artificial neural networks for atomistic modeling of materials Nat. Commun. 10 2339 doi: 10.1038/s41467-019-10343-5
|
| [32] |
Bartk A P, Payne M C, Kondor R, Csnyi G 2010 Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons Phys. Rev. Lett. 104 136403 doi: 10.1103/PhysRevLett.104.136403
|
| [33] |
Dragoni D, Daff T D, Csnyi G, Marzari N 2018 Achieving DFT accuracy with a machine-learning interatomic potential: thermomechanics and defects in bcc ferromagnetic iron Phys. Rev. Mater. 2 013808 doi: 10.1103/PhysRevMaterials.2.013808
|
| [34] |
Bartk A P, Kermode J, Bernstein N, Csnyi G 2018 Machine learning a general-purpose interatomic potential for silicon Phys. Rev. X 8 041048 doi: 10.1103/PhysRevX.8.041048
|
| [35] |
Deringer V L, Bartk A P, Bernstein N, Wilkins D M, Ceriotti M, Csnyi G 2021 Gaussian process regression for materials and molecules Chem. Rev. 121 10073 doi: 10.1021/acs.chemrev.1c00022
|
| [36] |
Shapeev A V 2016 Moment tensor potentials: a class of systematically improvable interatomic potentials Multiscale Model. Simul. 14 1153 doi: 10.1137/15M1054183
|
| [37] |
Podryabinkin E V, Shapeev A V 2017 Active learning of linearly parametrized interatomic potentials Comput. Mater. Sci. 140 171 doi: 10.1016/j.commatsci.2017.08.031
|
| [38] |
Podryabinkin E V, Tikhonov E V, Shapeev A V, Oganov A R 2019 Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning Phys. Rev. B 99 064114 doi: 10.1103/PhysRevB.99.064114
|
| [39] |
Chen C, Deng Z, Tran R, Tang H, Chu I-H, Ong S P 2017 Accurate force field for molybdenum by machine learning large materials data Phys. Rev. Mater. 1 043603 doi: 10.1103/PhysRevMaterials.1.043603
|
| [40] |
Li X-G, Hu C, Chen C, Deng Z, Luo J, Ong S P 2018 Quantum-accurate spectral neighbor analysis potential models for Ni-Mo binary alloys and fcc metals Phys. Rev. B 98 094104 doi: 10.1103/PhysRevB.98.094104
|
| [41] |
Deng Z, Chen C, Li X-G, Ong S P 2019 An electrostatic spectral neighbor analysis potential for lithium nitride npj Comput. Mater. 5 75 doi: 10.1038/s41524-019-0212-1
|
| [42] |
Sauceda H E, Chmiela S, Poltavsky I, Mller K-R, Tkatchenko A 2019 Molecular force fields with gradient-domain machine learning: construction and application to dynamics of small molecules with coupled cluster forces J. Chem. Phys. 150 114102 doi: 10.1063/1.5078687
|
| [43] |
Chmiela S, Sauceda H E, Poltavsky I, Mller K-R, Tkatchenko A 2019 sGDML: constructing accurate and data efficient molecular force fields using machine learning Comput. Phys. Commun. 240 38 doi: 10.1016/j.cpc.2019.02.007
|
| [44] |
Unke O T, Chmiela S, Sauceda H E, Gastegger M, Poltavsky I, Schtt K T, Tkatchenko A, Mller K-R 2021 Machine learning force fields Chem. Rev. 121 10142 doi: 10.1021/acs.chemrev.0c01111
|
| [45] |
Zuo Y, et al 2020 Performance and cost assessment of machine learning interatomic potentials J. Phys. Chem. A 124 731 doi: 10.1021/acs.jpca.9b08723
|
| [46] |
Han J, Zhang L, Car R, E W 2017 Deep potential: a general representation of a many-body potential energy surface arXiv:1707.01478 [physics.comp-ph])
|
| [47] |
Han J, Zhang L, Car R, E W 2018 Deep potential: a general representation of a many-body potential energy surface Commun. Comput. Phys. 23 629 doi: 10.4208/cicp.OA-2017-0213
|
| [48] |
Zhang L, Han J, Wang H, Car R, E W 2018 Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics Phys. Rev. Lett. 120 143001 doi: 10.1103/PhysRevLett.120.143001
|
| [49] |
Wang H, Zhang L, Han J, E W 2018 Deepmd-kit: a deep learning package for many-body potential energy representation and molecular dynamics Comput. Phys. Commun. 228 178 doi: 10.1016/j.cpc.2018.03.016
|
| [50] |
Jia W, Wang H, Chen M, Lu D, Lin L, Car R, E W, Zhang L 2020 Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning SC20: Int. Conf. for High Performance Computing, Networking, Storage and Analysis pp 1-14
|
| [51] |
Artrith N, Morawietz T, Behler J 2011 High-dimensional neural-network potentials for multicomponent systems: applications to zinc oxide Phys. Rev. B 83 153101 doi: 10.1103/PhysRevB.83.153101
|
| [52] |
Bereau T, Andrienko D, von Lilienfeld O A 2015 Transferable atomic multipole machine learning models for small organic molecules J. Chem. Theory Comput. 11 3225 doi: 10.1021/acs.jctc.5b00301
|
| [53] |
Bereau T, DiStasio R A, Tkatchenko A, von Lilienfeld O A 2018 Non-covalent interactions across organic and biological subsets of chemical space: Physics-based potentials parametrized from machine learning J. Chem. Phys. 148 241706 doi: 10.1063/1.5009502
|
| [54] |
Nebgen B, Lubbers N, Smith J S, Sifain A, Lokhov A, Isayev O, Roitberg A, Barros K, Tretiak S 2018 Transferable molecular charge assignment using deep neural networks arXiv:1803.04395 [physics.chem-ph])
|
| [55] |
Sifain A E, Lubbers N, Nebgen B T, Smith J S, Lokhov A Y, Isayev O, Roitberg A E, Barros K, Tretiak S 2018 Discovering a transferable charge assignment model using machine learning J. Phys. Chem. Lett. 9 4495 doi: 10.1021/acs.jpclett.8b01939
|
| [56] |
Ko T W, Finkler J A, Goedecker S, Behler J 2021 A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer Nat. Commun. 12 398 doi: 10.1038/s41467-020-20427-2
|
| [57] |
Ko T W, Finkler J A, Goedecker S, Behler J 2021 General-purpose machine learning potentials capturing nonlocal charge transfer Acc. Chem. Res. 54 808 doi: 10.1021/acs.accounts.0c00689
|
| [58] |
Grisafi A, Ceriotti M 2019 Incorporating long-range physics in atomic-scale machine learning J. Chem. Phys. 151 204105 doi: 10.1063/1.5128375
|
| [59] |
Grisafi A, Nigam J, Ceriotti M 2021 Multi-scale approach for the prediction of atomic scale properties Chem. Sci. 12 2078 doi: 10.1039/D0SC04934D
|
| [60] |
Frenkel D, Smit B 2002 Understanding Molecular Simulation From Algorithms to ApplicationsNew YorkAcademic
|
| [61] |
He K, Zhang X, Ren S, Sun J 2016 Deep residual learning for image recognition Proc. Conf. on Computer Vision and Pattern Recognition (CVPR)
|
| [62] |
Barron A 1993 Universal approximation bounds for superpositions of a sigmoidal function IEEE Trans. Inf. Theory 39 930 doi: 10.1109/18.256500
|
| [63] |
Barron A R 1994 Approximation and estimation bounds for artificial neural networks Mach. Learn. 14 115 doi: 10.1007/BF00993164
|
| [64] |
Liang S, Srikant R 2017 Why deep neural networks for function approximation? arXiv:1610.04161 [cs.LG])
|
| [65] |
Telgarsky M 2016 benefits of depth in neural networks 29th Conf. on Learning Theory (Proc. Machine Learning Research) (PMLR)vol 49ed(V Feldman, A Rakhlin and O Shamir)(New York: Columbia University) 1517-39
|
| [66] |
Yarotsky D 2017 Error bounds for approximations with deep relu networks Neural Netw. 94 103 doi: 10.1016/j.neunet.2017.07.002
|
| [67] |
Lu J, Shen Z, Yang H, Zhang S 2021 Deep network approximation for smooth functions SIAM J. Math. Anal. 53 5465 doi: 10.1137/20M134695X
|
| [68] |
E W, Ma C, Wu L 2019 A priori estimates of the population risk for two-layer neural networks Commun. Math. Sci. 17 1407-25 doi: 10.4310/CMS.2019.v17.n5.a11
|
| [69] |
E W, Ma C, Wu L 2022 The barron space and the flow-induced function spaces for neural network models Constructive Approx. 55 369-406 doi: 10.1007/s00365-021-09549-y
|
| [70] |
Zhang L, Han J, Wang H, Saidi W, Car R, E W 2018 End-to-end symmetry preserving inter-atomic potential energy model for finite and extended systems Advances in Neural Information Processing Systemsvol 31Bengio S, Wallach H, Larochelle H, Grauman K, Cesa-Bianchi N, Garnett R 2018 (Curran Associates, Inc.) pp 4436-46
|
| [71] |
Kresse G, Furthmller J 1996 Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set Comput. Mater. Sci. 6 15 doi: 10.1016/0927-0256(96)00008-0
|
| [72] |
Kresse G, Furthmller J 1996 Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set Phys. Rev. B 54 11169 doi: 10.1103/PhysRevB.54.11169
|
| [73] |
Giannozzi P, et al 2009 QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials J. Phys.: Condens. Matter. 21 395502 doi: 10.1088/0953-8984/21/39/395502
|
| [74] |
Chen M, Guo G-C, He L 2010 Systematically improvable optimized atomic basis sets for ab initio calculations J. Phys.: Condens. Matter. 22 445501 doi: 10.1088/0953-8984/22/44/445501
|
| [75] |
Perdew J P, Schmidt K 2001 Jacob’s ladder of density functional approximations for the exchange-correlation energy Conf. Proc.vol 577 p 1
|
| [76] |
Mller C, Plesset M S 1934 Note on an approximation treatment for many-electron systems Phys. Rev. 46 618 doi: 10.1103/PhysRev.46.618
|
| [77] |
ek J 1996 On the correlation problem in atomic and molecular systems. Calculation of wavefunction components in ursell-type expansion using quantum-field theoretical methods J. Chem. Phys. 45 4256 doi: 10.1063/1.1727484
|
| [78] |
Fano U 1961 Effects of configuration interaction on intensities and phase shifts Phys. Rev. 124 1866 doi: 10.1103/PhysRev.124.1866
|
| [79] |
Deaven D M, Ho K M 1995 Molecular geometry optimization with a genetic algorithm Phys. Rev. Lett. 75 288 doi: 10.1103/PhysRevLett.75.288
|
| [80] |
Glass C W, Oganov A R, Hansen N 2006 USPEX-evolutionary crystal structure prediction Comput. Phys. Commun. 175 713 doi: 10.1016/j.cpc.2006.07.020
|
| [81] |
Laio A, Parrinello M 2002 Escaping free-energy minima Proc. Natl Acad. Sci. 99 12562 doi: 10.1073/pnas.202427399
|
| [82] |
Cohn D, Atlas L, Ladner R 1994 Improving generalization with active learning Mach. Learn. 15 201 doi: 10.1007/BF00993277
|
| [83] |
Zhang L, Lin D-Y, Wang H, Car R, E W 2019 Active learning of uniformly accurate interatomic potentials for materials simulation Phys. Rev. Mater. 3 023804 doi: 10.1103/PhysRevMaterials.3.023804
|
| [84] |
Zhang Y, Wang H, Chen W, Zeng J, Zhang L, Wang H, E W 2020 DP-GEN: a concurrent learning platform for the generation of reliable deep learning based potential energy models Comput. Phys. Commun. 253 107206 doi: 10.1016/j.cpc.2020.107206
|
| [85] |
Plimpton S 1995 Fast parallel algorithms for short-range molecular dynamics J. Comput. Phys. 117 1 doi: 10.1006/jcph.1995.1039
|
| [86] |
Larsen A H, et al 2017 The atomic simulation environmenta python library for working with atoms J. Phys.: Condens. Matter. 29 273002 doi: 10.1088/1361-648x/aa680e
|
| [87] |
Ceriotti M, More J, Manolopoulos D E 2014 i-PI: a python interface for ab initio path integral molecular dynamics simulations Comput. Phys. Commun. 185 1019 doi: 10.1016/j.cpc.2013.10.027
|
| [88] |
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark A E, Berendsen H J C 2005 Gromacs: fast, flexible and free J. Comput. Chem. 26 1701 doi: 10.1002/jcc.20291
|
| [89] |
Gaussian 16 Revision C.01 2016 Gaussian Inc. Wallingford CT
|
| [90] |
Soler J M, Artacho E, Gale J D, Garca A, Junquera J, Ordejn P, Snchez-Portal D 2002 The SIESTA method for ab initio order-N materials simulation J. Phys.: Condens. Matter. 14 2745 doi: 10.1088/0953-8984/14/11/302
|
| [91] |
Khne T D, et al 2020 Cp2k: an electronic structure and molecular dynamics software packagequickstep: efficient and accurate electronic structure calculations J. Chem. Phys. 152 194103 doi: 10.1063/5.0007045
|
| [92] |
Blum V, Gehrke R, Hanke F, Havu P, Havu V, Ren X, Reuter K, Scheffler M 2009 Ab initio molecular simulations with numeric atom-centered orbitals Comput. Phys. Commun. 180 2175 doi: 10.1016/j.cpc.2009.06.022
|
| [93] |
Case D A, Cheatham III T E, Darden T, Gohlke H, Luo R, Merz Jr. K M, Onufriev A, Simmerling C, Wang B, Woods R J 2005 The amber biomolecular simulation programs J. Comput. Chem. 26 1668 doi: 10.1002/jcc.20290
|
| [94] |
Abadi M, et al 2015 TensorFlow: large-scale machine learning on heterogeneous systems software available fromwww.tensorflow.org/
|
| [95] |
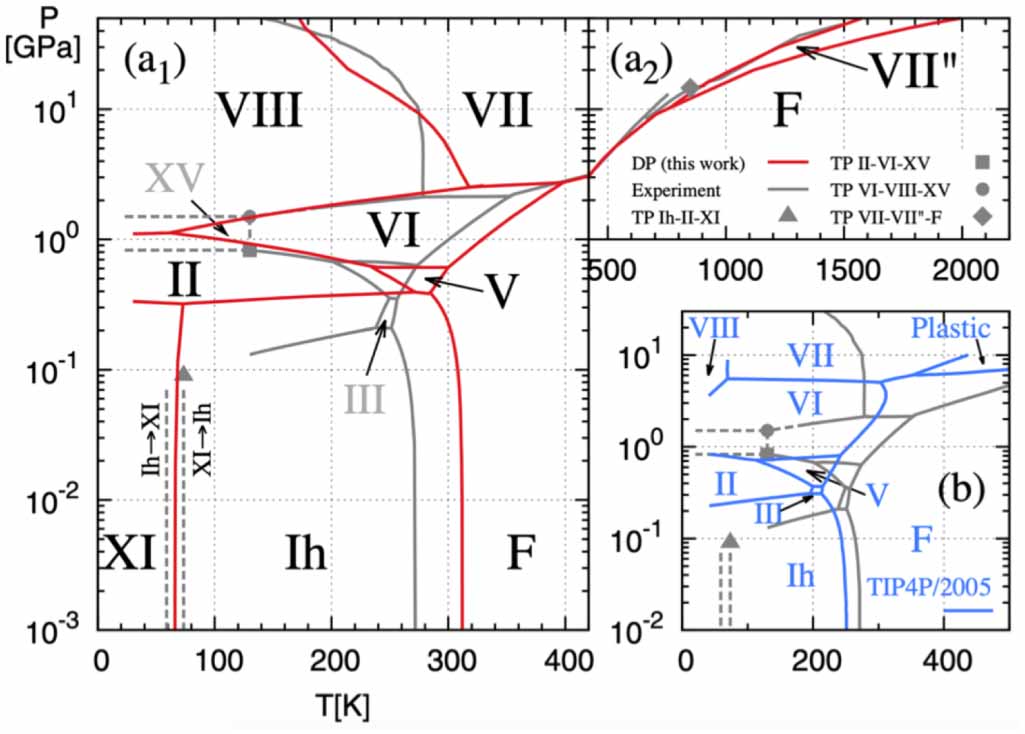
Zhang L, Wang H, Car R, E W 2021 Phase diagram of a deep potential water model Phys. Rev. Lett. 126 236001 doi: 10.1103/PhysRevLett.126.236001
|
| [96] |
Schimka L, Gaudoin R, Klime J, Marsman M, Kresse G 2013 Lattice constants and cohesive energies of alkali, alkaline-earth and transition metals: random phase approximation and density functional theory results Phys. Rev. B 87 214102 doi: 10.1103/PhysRevB.87.214102
|
| [97] |
Kittel C 2005 Introduction to Solid State Physics8th ednNew YorkWiley
|
| [98] |
Yang M, Karmakar T, Parrinello M 2021 Liquid-liquid critical point in phosphorus Phys. Rev. Lett. 127 080603 doi: 10.1103/PhysRevLett.127.080603
|
| [99] |
Yang M, Bonati L, Polino D, Parrinello M 2022 Using metadynamics to build neural network potentials for reactive events: the case of urea decomposition in water Catal. Today 387 143 doi: 10.1016/j.cattod.2021.03.018
|
| [100] |
Jiang W, Zhang Y, Zhang L, Wang H 2021 Accurate Deep Potential model for the Al-Cu-Mg alloy in the full concentration space Chin. Phys. B 30 050706 doi: 10.1088/1674-1056/abf134
|
| [101] |
Wen T, Wang R, Zhu L, Zhang L, Wang H, Srolovitz D J, Wu Z 2021 Specialising neural network potentials for accurate properties and application to the mechanical response of titanium npj Comput. Mater. 7 206 doi: 10.1038/s41524-021-00661-y
|
| [102] |
Wang X, Wang Y, Zhang L, Dai F, Wang H 2021 A tungsten deep potential with high accuracy and generalization ability based on a newly designed three-body embedding formalism arXiv:2111.04281 [cond-mat.mtrl-sci])
|
| [103] |
Wang Y, Zhang L, Xu B, Wang X, Wang H 2022 A generalizable machine learning potential of Ag-Au nanoalloys and its application to surface reconstruction, segregation and diffusion Modelling Simul. Mater. Sci. Eng. 30 025003 doi: 10.1088/1361-651X/ac4002
|
| [104] |
Fu B, Sun Y, Zhang L, Wang H, Xu B 2021 Deep learning inter-atomic potential for thermal and phonon behaviour of silicon carbide with quantum accuracy arXiv:2110.10843 [cond-mat.mtrl-sci])
|
| [105] |
Huang J, Zhang L, Wang H, Zhao J, Cheng J, E W 2021 Deep potential generation scheme and simulation protocol for the Li10GeP2S12-type superionic conductors J. Chem. Phys. 154 094703 doi: 10.1063/5.0041849
|
| [106] |
Lu D, Wang H, Chen M, Lin L, Car R, E W, Jia W, Zhang L 2021 86 PFLOPS deep potential molecular dynamics simulation of 100 million atoms with ab initio accuracy Comput. Phys. Commun. 259 107624 doi: 10.1016/j.cpc.2020.107624
|
| [107] |
Lu D, Jiang W, Chen Y, Zhang L, Jia W, Wang H, Chen M 2021 DP train, then DP compress: model compression in deep potential molecular dynamics arXiv:2107.02103 [physics.comp-ph])
|
| [108] |
Mendelev M, Underwood T, Ackland G 2016 Development of an interatomic potential for the simulation of defects, plasticity and phase transformations in titanium J. Chem. Phys. 145 154102 doi: 10.1063/1.4964654
|
| [109] |
Hennig R, Lenosky T, Trinkle D, Rudin S, Wilkins J W 2008 Classical potential describes martensitic phase transformations between the
|
| [110] |
Vtek V 1968 Intrinsic stacking faults in body-centred cubic crystals Phil. Mag. 18 773 doi: 10.1080/14786436808227500
|
| [111] |
Ko W-S, Grabowski B, Neugebauer J 2015 Development and application of a Ni-Ti interatomic potential with high predictive accuracy of the martensitic phase transition Phys. Rev. B 92 134107 doi: 10.1103/PhysRevB.92.134107
|
| [112] |
Dickel D, Barrett C, Carino R, Baskes M, Horstemeyer M 2018 Mechanical instabilities in the modeling of phase transitions of titanium Modelling Simul. Mater. Sci. Eng. 26 065002 doi: 10.1088/1361-651X/aac95d
|
| [113] |
Clouet E, Caillard D, Chaari N, Onimus F, Rodney D 2015 Dislocation locking versus easy glide in titanium and zirconium Nat. Mater. 14 931 doi: 10.1038/nmat4340
|
| [114] |
Wang H, Guo X, Zhang L, Wang H, Xue J 2019 Deep learning inter-atomic potential model for accurate irradiation damage simulations Appl. Phys. Lett. 114 244101 doi: 10.1063/1.5098061
|
| [115] |
Zeng Q, Yu X, Yao Y, Gao T, Chen B, Zhang S, Kang D, Wang H, Dai J 2021 Ab initio validation on the connection between atomistic and hydrodynamic description to unravel the ion dynamics of warm dense matter Phys. Rev. Res. 3 033116 doi: 10.1103/PhysRevResearch.3.033116
|
| [116] |
Liu Q, Lu D, Chen M 2020 Structure and dynamics of warm dense aluminum: a molecular dynamics study with density functional theory and deep potential J. Phys.: Condens. Matter. 32 144002 doi: 10.1088/1361-648x/ab5890
|
| [117] |
Liu Q, Li J, Chen M 2021 Thermal transport by electrons and ions in warm dense aluminum: a combined density functional theory and deep potential study Matter Radiat. Extremes 6 026902 doi: 10.1063/5.0030123
|
| [118] |
Cheng Y, et al 2021 Deep-learning potential method to simulate shear viscosity of liquid aluminum at high temperature and high pressure by molecular dynamics AIP Adv. 11 015043 doi: 10.1063/5.0036298
|
| [119] |
Andolina C M, Bon M, Passerone D, Saidi W A 2021 Robust, multi-length-scale, machine learning potential for Ag-Au bimetallic alloys from clusters to bulk materials J. Phys. Chem. C 125 17438 doi: 10.1021/acs.jpcc.1c04403
|
| [120] |
Chen B, Zeng Q, Wang H, Zhang S, Kang D, Lu D, Dai J 2021 Atomistic mechanism of phase transition in shock compressed gold revealed by deep potential arXiv:2006.13136 [cond-mat.mtrl-sci])
|
| [121] |
Jiao J 2021 Self-healing mechanism of lithium in lithium metal batteries arXiv:2106.10979 [cond-mat.mtrl-sci])
|
| [122] |
Zhang Y, Gao C, Liu Q, Zhang L, Wang H, Chen M 2020 Warm dense matter simulation via electron temperature dependent deep potential molecular dynamics Phys. Plasmas 27 122704 doi: 10.1063/5.0023265
|
| [123] |
Niu H, Bonati L, Piaggi P M, Parrinello M 2020 Ab initio phase diagram and nucleation of gallium Nat. Commun. 11 1 doi: 10.1038/s41467-020-16372-9
|
| [124] |
Shi M, Li J, Tao M, Zhang X, Liu J 2021 Artificial intelligence model for efficient simulation of monatomic phase change material antimony Mater. Sci. Semicond. Process. 136 106146 doi: 10.1016/j.mssp.2021.106146
|
| [125] |
Wang J, Shen H, Yang R, Xie K, Zhang C, Chen L, Ho K-M, Wang C-Z, Wang S 2022 A deep learning interatomic potential developed for atomistic simulation of carbon materials Carbon 186 1 doi: 10.1016/j.carbon.2021.09.062
|
| [126] |
Bonati L, Parrinello M 2018 Silicon liquid structure and crystal nucleation from ab Initio deep metadynamics Phys. Rev. Lett. 121 265701 doi: 10.1103/PhysRevLett.121.265701
|
| [127] |
Li R, Lee E, Luo T 2020 A unified deep neural network potential capable of predicting thermal conductivity of silicon in different phases Mater. Today Phys. 12 100181 doi: 10.1016/j.mtphys.2020.100181
|
| [128] |
Wang H, Zhang Y, Zhang L, Wang H 2020 Crystal structure prediction of binary alloys via deep potential Front. Chem. 8 895 doi: 10.3389/fchem.2020.589795
|
| [129] |
Andolina C M, Wright J G, Das N, Saidi W A 2021 Improved Al-Mg alloy surface segregation predictions with a machine learning atomistic potential Phys. Rev. Mater. 5 083804 doi: 10.1103/PhysRevMaterials.5.083804
|
| [130] |
Bourgeois L, Zhang Y, Zhang Z, Chen Y, Medhekar N V 2020 Transforming solid-state precipitates via excess vacancies Nat. Commun. 11 1 doi: 10.1038/s41467-020-15087-1
|
| [131] |
Cheng B, Zhao X, Zhang Y, Chen H, Polmear I, Nie J-F 2020 Co-segregation of Mg and Zn atoms at the planar
|
| [132] |
Ryltsev R E, Chtchelkatchev N M 2021 Deep machine learning potentials for multicomponent metallic melts: development, predictability and compositional transferability arXiv:2110.14006 [cond-mat.mtrl-sci])
|
| [133] |
Wen T, et al 2019 Development of a deep machine learning interatomic potential for metalloid-containing Pd-Si compounds Phys. Rev. B 100 174101 doi: 10.1103/PhysRevB.100.174101
|
| [134] |
Wang Q, Zhai B, Wang H P, Wei B 2021 Atomic structure of liquid refractory Nb5Si3 intermetallic compound alloy based upon deep neural network potential J. Appl. Phys. 130 185103 doi: 10.1063/5.0067157
|
| [135] |
Guo Y, et al 2019 Bergman-type medium range order in amorphous Zr77Rh23 alloy studied by ab initio molecular dynamics simulations J. Alloys Compd. 790 675 doi: 10.1016/j.jallcom.2019.03.197
|
| [136] |
Guo Y, et al 2019 Temperature dependence of structural, dynamical and electronic properties of amorphous Bi2Te3: an ab initio study New J. Phys. 21 093062 doi: 10.1088/1367-2630/ab4535
|
| [137] |
Tang L, Yang Z J, Wen T Q, Ho K M, Kramer M J, Wang C Z 2020 Development of interatomic potential for Al-Tb alloys using a deep neural network learning method Phys. Chem. Chem. Phys. 22 18467 doi: 10.1039/D0CP01689F
|
| [138] |
Tang L, Yang Z, Wen T, Ho K M, Kramer M J, Wang C Z 2021 Short- and medium-range orders in Al90Tb10 glass and their relation to the structures of competing crystalline phases Acta Mater. 204 116513 doi: 10.1016/j.actamat.2020.116513
|
| [139] |
Han I, McKeown J T, Tang L, Wang C-Z, Parsamehr H, Xi Z, Lu Y-R, Kramer M J, Shahani A J 2020 Dynamic observation of dendritic quasicrystal growth upon laser-induced solid-state transformation Phys. Rev. Lett. 125 195503 doi: 10.1103/PhysRevLett.125.195503
|
| [140] |
Tang L, Ho K M, Wang C Z 2021 Molecular dynamics simulation of metallic Al-Ce liquids using a neural network machine learning interatomic potential J. Chem. Phys. 155 194503 doi: 10.1063/5.0066061
|
| [141] |
Daniels C L, Liu D-J, Adamson M A S, Knobeloch M, Vela J 2021 Azo(xy) vs aniline selectivity in catalytic nitroarene reduction by intermetallics: experiments and simulations J. Phys. Chem. C 125 24440 doi: 10.1021/acs.jpcc.1c08569
|
| [142] |
Zhang C, Sun Y, Wang H-D, Zhang F, Wen T-Q, Ho K-M, Wang C-Z 2021 Crystallization of the P3Sn4 phase upon cooling P2Sn5 liquid by molecular dynamics simulation using a machine learning interatomic potential J. Phys. Chem. C 125 3127 doi: 10.1021/acs.jpcc.0c08873
|
| [143] |
Balyakin I A, Rempel S V, Ryltsev R E, Rempel A A 2020 Deep machine learning interatomic potential for liquid silica Phys. Rev. E 102 052125 doi: 10.1103/PhysRevE.102.052125
|
| [144] |
Deng J, Stixrude L 2021 Thermal conductivity of silicate liquid determined by machine learning potentials Geophys. Res. Lett. 48 e2021GL093806 doi: 10.1029/2021GL093806
|
| [145] |
Luo H, Karki B B, Ghosh D B, Bao H 2021 Anomalous behavior of viscosity and electrical conductivity of MgSiO3 melt at mantle conditions Geophys. Res. Lett. 48 e2021GL093573 doi: 10.1029/2021GL093573
|
| [146] |
Luo H, Karki B B, Ghosh D B, Bao H 2021 Deep neural network potentials for diffusional lithium isotope fractionation in silicate melts Geochim. Cosmochim. Acta 303 38 doi: 10.1016/j.gca.2021.03.031
|
| [147] |
Chen W, Li L-S 2021 The study of the optical phonon frequency of 3C-SiC by molecular dynamics simulations with deep neural network potential J. Appl. Phys. 129 244104 doi: 10.1063/5.0049464
|
| [148] |
An Q 2021 Mitigating amorphization in superhard boron carbide by microalloying-induced stacking fault formation Phys. Rev. Mater. 5 103602 doi: 10.1103/PhysRevMaterials.5.103602
|
| [149] |
Rodriguez A, Lam S, Hu M 2021 Thermodynamic and transport properties of LiF and FLiBe molten salts with deep learning potentials ACS Appl. Mater. Interfaces 13 55367-79 doi: 10.1021/acsami.1c17942
|
| [150] |
Liang W, Lu G, Yu J 2021 Theoretical prediction on the local structure and transport properties of molten alkali chlorides by deep potentials J. Mater. Sci. Technol. 75 78 doi: 10.1016/j.jmst.2020.09.040
|
| [151] |
Liang W, Lu G, Yu J 2020 Molecular dynamics simulations of molten magnesium chloride using machine-learning-based deep potential Adv. Theory Simul. 3 2000180 doi: 10.1002/adts.202000180
|
| [152] |
Pan G, Chen P, Yan H, Lu Y 2020 A DFT accurate machine learning description of molten ZnCl2 and its mixtures: 1. Potential development and properties prediction of molten ZnCl2 Comput. Mater. Sci. 185 109955 doi: 10.1016/j.commatsci.2020.109955
|
| [153] |
Pan G, Ding J, Du Y, Lee D-J, Lu Y 2021 A DFT accurate machine learning description of molten ZnCl2 and its mixtures: 2. Potential development and properties prediction of ZnCl2-NaCl-KCl ternary salt for CSP Comput. Mater. Sci. 187 110055 doi: 10.1016/j.commatsci.2020.110055
|
| [154] |
Liang W, Lu G, Yu J 2021 Machine-learning-driven simulations on microstructure and thermophysical properties of MgCl2-KCl eutectic ACS Appl. Mater. Interfaces 13 4034 doi: 10.1021/acsami.0c20665
|
| [155] |
Bu M, Liang W, Lu G, Yu J 2021 Local structure elucidation and properties prediction on KCl-CaCl2 molten salt: a deep potential molecular dynamics study Sol. Energy Mater. Sol. Cells 232 111346 doi: 10.1016/j.solmat.2021.111346
|
| [156] |
Zhao J, Liang W, Lu G 2021 Theoretical prediction on the redox potentials of rare-earth ions by deep potentials Ionics 27 2079 doi: 10.1007/s11581-021-03988-0
|
| [157] |
Zhang J, Fuller J, An Q 2021 Coordination and thermophysical properties of transition metal chlorocomplexes in LiCl-KCl eutectic J. Phys. Chem. B 125 8876 doi: 10.1021/acs.jpcb.1c03748
|
| [158] |
Xu N, Shi Y, He Y, Shao Q 2020 A deep-learning potential for crystalline and amorphous Li-Si alloys J. Phys. Chem. C 124 16278 doi: 10.1021/acs.jpcc.0c03333
|
| [159] |
Marcolongo A, Binninger T, Zipoli F, Laino T 2019 Simulating diffusion properties of solid-state electrolytes via a neural network potential: performance and training scheme arXiv:1910.10090 [physics.comp-ph])
|
| [160] |
Gupta M K, Ding J, Osti N C, Abernathy D L, Arnold W, Wang H, Hood Z, Delaire O 2021 Fast Na diffusion and anharmonic phonon dynamics in superionic Na3PS4 Energy Environ. Sci. 14 6554 doi: 10.1039/D1EE01509E
|
| [161] |
Li H-X, Zhou X-Y, Wang Y-C, Jiang H 2021 Theoretical study of Na+ transport in the solid-state electrolyte Na3OBr based on deep potential molecular dynamics Inorg. Chem. Front. 8 425 doi: 10.1039/D0QI00921K
|
| [162] |
Lin M, Liu X, Xiang Y, Wang F, Liu Y, Fu R, Cheng J, Yang Y 2021 Unravelling the fast alkali-ion dynamics in paramagnetic battery materials combined with NMR and deep-potential molecular dynamics simulation Angew. Chem., Int. Ed. 60 12547 doi: 10.1002/anie.202102740
|
| [163] |
Calegari Andrade M F, Selloni A 2020 Structure of disordered TiO2 phases from ab initio based deep neural network simulations Phys. Rev. Mater. 4 113803 doi: 10.1103/PhysRevMaterials.4.113803
|
| [164] |
Li R, Liu Z, Rohskopf A, Gordiz K, Henry A, Lee E, Luo T 2020 A deep neural network interatomic potential for studying thermal conductivity of
|
| [165] |
Wu J, Zhang Y, Zhang L, Liu S 2021 Deep learning of accurate force field of ferroelectric HfO2 Phys. Rev. B 103 024108 doi: 10.1103/PhysRevB.103.024108
|
| [166] |
Balyakin I, Sadovnikov S 2022 Deep learning potential for superionic phase of Ag2S Comput. Mater. Sci. 202 110963 doi: 10.1016/j.commatsci.2021.110963
|
| [167] |
Wang H, Guo X, Xue J 2020 Deep-learning interatomic potential for irradiation damage simulations in MoS2 with ab initial accuracy arXiv:2010.09547 [cond-mat.mtrl-sci])
|
| [168] |
Guo D, Li C, Li K, Shao B, Chen D, Ma Y, Sun J, Cao X, Zeng W, Chang X 2021 The thermoelectric performance of new structure SnSe studied by quotient graph and deep learning potential Mater. Today Energy 20 100665 doi: 10.1016/j.mtener.2021.100665
|
| [169] |
Dai F-Z, Wen B, Xiang H, Zhou Y 2020 Grain boundary strengthening in ZrB2 by segregation of W: atomistic simulations with deep learning potential J. Eur. Ceram. Soc. 40 5029 doi: 10.1016/j.jeurceramsoc.2020.06.007
|
| [170] |
Dai F-Z, Wen B, Sun Y, Xiang H, Zhou Y 2020 Theoretical prediction on thermal and mechanical properties of high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C by deep learning potential J. Mater. Sci. Technol. 43 168 doi: 10.1016/j.jmst.2020.01.005
|
| [171] |
Dai F-Z, Sun Y, Wen B, Xiang H, Zhou Y 2021 Temperature dependent thermal and elastic properties of high entropy (Ti0.2Zr0.2Hf0.2Nb0.2Ta0.2)B2: molecular dynamics simulation by deep learning potential J. Mater. Sci. Technol. 72 8 doi: 10.1016/j.jmst.2020.07.014
|
| [172] |
Ko H-Y, Zhang L, Santra B, Wang H, E W, DiStasio Jr R A, Car R 2019 Isotope effects in liquid water via deep potential molecular dynamics Mol. Phys. 117 3269 doi: 10.1080/00268976.2019.1652366
|
| [173] |
Sommers G M, Calegari Andrade M F, Zhang L, Wang H, Car R 2020 Raman spectrum and polarizability of liquid water from deep neural networks Phys. Chem. Chem. Phys. 22 10592 doi: 10.1039/D0CP01893G
|
| [174] |
Zhang C, Zhang L, Xu J, Tang F, Santra B, Wu X 2020 Isotope effects in x-ray absorption spectra of liquid water Phys. Rev. B 102 115155 doi: 10.1103/PhysRevB.102.115155
|
| [175] |
Gartner T E, Zhang L, Piaggi P M, Car R, Panagiotopoulos A Z, Debenedetti P G 2020 Signatures of a liquid-liquid transition in an ab initio deep neural network model for water Proc. Natl Acad. Sci. 117 26040 doi: 10.1073/pnas.2015440117
|
| [176] |
Andreani C, Romanelli G, Parmentier A, Senesi R, Kolesnikov A I, Ko H-Y, Calegari Andrade M F, Car R 2020 Hydrogen dynamics in supercritical water probed by neutron scattering and computer simulations J. Phys. Chem. Lett. 11 9461 doi: 10.1021/acs.jpclett.0c02547
|
| [177] |
Xu J, Zhang C, Zhang L, Chen M, Santra B, Wu X 2020 Isotope effects in molecular structures and electronic properties of liquid water via deep potential molecular dynamics based on the SCAN functional Phys. Rev. B 102 214113 doi: 10.1103/PhysRevB.102.214113
|
| [178] |
Piaggi P M, Panagiotopoulos A Z, Debenedetti P G, Car R 2021 Phase equilibrium of water with hexagonal and cubic ice using the SCAN functional J. Chem. Theory Comput. 17 3065 doi: 10.1021/acs.jctc.1c00041
|
| [179] |
Tisi D, Zhang L, Bertossa R, Wang H, Car R, Baroni S 2021 Heat transport in liquid water from first-principles and deep-neural-network simulations arXiv:2108.10850 [cond-mat.mtrl-sci])
|
| [180] |
Zhang C, Tang F, Chen M, Xu J, Zhang L, Qiu D Y, Perdew J P, Klein M L, Wu X 2021 Modeling liquid water by climbing up Jacob’s ladder in density functional theory facilitated by using deep neural network potentials J. Phys. Chem. B 125 11444 doi: 10.1021/acs.jpcb.1c03884
|
| [181] |
Torres A, Pedroza L S, Fernandez-Serra M, Rocha A R 2021 Using neural network force fields to ascertain the quality of ab initio simulations of liquid water J. Phys. Chem. B 125 10772 doi: 10.1021/acs.jpcb.1c04372
|
| [182] |
Shi Y, Doyle C C, Beck T L 2021 Condensed phase water molecular multipole moments from deep neural network models trained on ab initio simulation data J. Phys. Chem. Lett. 12 10310 doi: 10.1021/acs.jpclett.1c02328
|
| [183] |
Calio P B, Li C, Voth G A 2021 Resolving the structural debate for the hydrated excess proton in water J. Am. Chem. Soc. 143 18672 doi: 10.1021/jacs.1c08552
|
| [184] |
Xu M, Zhu T, Zhang J Z H 2019 Molecular dynamics simulation of zinc ion in water with an ab initio based neural network potential J. Phys. Chem. A 123 6587 doi: 10.1021/acs.jpca.9b04087
|
| [185] |
Niblett S P, Galib M, Limmer D T 2021 Learning intermolecular forces at liquid-vapor interfaces J. Chem. Phys. 155 164101 doi: 10.1063/5.0067565
|
| [186] |
Galib M, Limmer D T 2021 Reactive uptake of N2O5 by atmospheric aerosol is dominated by interfacial processes Science 371 921 doi: 10.1126/science.abd7716
|
| [187] |
Andrade M F C, Ko H-Y, Zhang L, Car R, Selloni A 2020 Free energy of proton transfer at the water-TiO2 interface from ab initio deep potential molecular dynamics Chem. Sci. 11 2335 doi: 10.1039/C9SC05116C
|
| [188] |
Piaggi P M, Car R 2021 Enhancing the formation of ionic defects to study the ice Ih/XI transition with molecular dynamics simulations Mol. Phys. 119 e1916634 doi: 10.1080/00268976.2021.1916634
|
| [189] |
Ye Q-J, Zhuang L, Li X-Z 2021 Dynamic nature of high-pressure ice VII Phys. Rev. Lett. 126 185501 doi: 10.1103/PhysRevLett.126.185501
|
| [190] |
Jiang S, Liu Y-R, Huang T, Feng Y-J, Wang C-Y, Wang Z-Q, Huang W 2021 Towards fully ab initio simulation of atmospheric aerosol nucleation arXiv:2107.04802 [physics.atm-clus])
|
| [191] |
Zeng J, Zhang L, Wang H, Zhu T 2021 Exploring the chemical space of linear alkane pyrolysis via deep potential generator Energy Fuels 35 762 doi: 10.1021/acs.energyfuels.0c03211
|
| [192] |
Chen W-K, Liu X-Y, Fang W-H, Dral P O, Cui G 2018 Deep learning for nonadiabatic excited-state dynamics J. Phys. Chem. Lett. 9 6702 doi: 10.1021/acs.jpclett.8b03026
|
| [193] |
Zhang L, Wang H, E W 2018 Reinforced dynamics for enhanced sampling in large atomic and molecular systems J. Chem. Phys. 148 124113 doi: 10.1063/1.5019675
|
| [194] |
Wang S, Ma Z, Pan W 2020 Data-driven coarse-grained modeling of polymers in solution with structural and dynamic properties conserved Soft Matter 16 8330 doi: 10.1039/D0SM01019G
|
| [195] |
Pan X, Yang J, Van R, Epifanovsky E, Ho J, Huang J, Pu J, Mei Y, Nam K, Shao Y 2021 Machine-learning-assisted free energy simulation of solution-phase and enzyme reactions J. Chem. Theory Comput. 17 5745 doi: 10.1021/acs.jctc.1c00565
|
| [196] |
Tuo P, Ye X B, Pan B C 2020 A machine learning based deep potential for seeking the low-lying candidates of Al clusters J. Chem. Phys. 152 114105 doi: 10.1063/5.0001491
|
| [197] |
Achar S K, Zhang L, Johnson J K 2021 Efficiently trained deep learning potential for graphane J. Phys. Chem. C 125 14874 doi: 10.1021/acs.jpcc.1c01411
|
| [198] |
Wu J, Bai L, Huang J, Ma L, Liu J, Liu S 2021 Accurate force field of two-dimensional ferroelectrics from deep learning Phys. Rev. B 104 174107 doi: 10.1103/PhysRevB.104.174107
|
| [199] |
Chen H, Chen J, Ning P, Chen X, Liang J, Yao X, Chen D, Qin L, Huang Y, Wen Z 2021 2D heterostructure of amorphous CoFeB coating black phosphorus nanosheets with optimal oxygen intermediate absorption for improved electrocatalytic water oxidation ACS Nano 15 12418 doi: 10.1021/acsnano.1c04715
|
| [200] |
Pascuet M, Fernndez J 2015 Atomic interaction of the MEAM type for the study of intermetallics in the Al-U alloy J. Nucl. Mater. 467 229 doi: 10.1016/j.jnucmat.2015.09.030
|
| [201] |
Jacobsen K W, Norskov J K, Puska M J 1987 Interatomic interactions in the effective-medium theory Phys. Rev. B 35 7423 doi: 10.1103/PhysRevB.35.7423
|
| [202] |
Jain A, et al 2013 Commentary: The materials project: a materials genome approach to accelerating materials innovation APL Mater. 1 011002 doi: 10.1063/1.4812323
|
| [203] |
Wang Y, Lv J, Zhu L, Ma Y 2012 Calypso: a method for crystal structure prediction Comput. Phys. Commun. 183 2063 doi: 10.1016/j.cpc.2012.05.008
|
| [204] |
Aragones J L, Conde M M, Noya E G, Vega C 2009 The phase diagram of water at high pressures as obtained by computer simulations of the tip4p/2005 model: the appearance of a plastic crystal phase Phys. Chem. Chem. Phys. 11 543 doi: 10.1039/B812834K
|
| [205] |
Poschmann M, Asta M, Chrzan D C 2018 Convergence of calculated dislocation core structures in hexagonal close packed titanium Modelling Simul. Mater. Sci. Eng. 26 014003 doi: 10.1088/1361-651X/aa9ba9
|
| [206] |
Queyroux J-A, et al 2020 Melting curve and isostructural solid transition in superionic ice Phys. Rev. Lett. 125 195501 doi: 10.1103/PhysRevLett.125.195501
|
| [207] |
Mishin Y 2021 Machine-learning interatomic potentials for materials science Acta Mater. 214 116980 doi: 10.1016/j.actamat.2021.116980
|
| [208] |
Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez A N, Kaiser L, Polosukhin I 2017 Attention is all you need available at: https://doi.org/10.48550/ARXIV.1706.03762
|
| [209] |
Devlin J, Chang M-W, Lee K, Toutanova K 2019 Bert: pre-training of deep bidirectional transformers for language understanding Proc. 2019th Conf. North American Chapter of the Association for Computational Linguistics: Human Language Technologies, (Long and Short Papers Vol 1)Minneapolis, MN(Association for Computational Linguistics) pp 4171-86
|
| [210] |
Brown T, et al 2020 Language models are few-shot learners Adv. Neural Inf. Process. Syst. 33 1877-901
|
| [211] |
Min B, Ross H, Sulem E, Veyseh A P B, Nguyen T H, Sainz O, Agirre E, Heinz I, Roth D 2021 Recent advances in natural language processing via large pre-trained language models: a survey available at:https://doi.org/10.48550/ARXIV.2111.01243
|

Figures(12) / Tables(2)



Copyright © 2021 Materials Futures Editorial Office
Supported by:
Beijing Renhe Information Technology Co., Ltd.




 DownLoad:
DownLoad: